HOME > SERVICES > EVALUATION AND MANAGEMENT OF HEART DISEASE > Cardiomyopathies >
Cardiomyopathies

Cardiomyopathy is a disease of the heart muscle that makes it harder for the heart to pump blood to the rest of the body. The various types of the disease have many causes, signs and symptoms as well as treatments. In most cases, cardiomyopathy causes the heart muscle to become enlarged, thick or rigid. In rare instances, diseased heart muscle tissue is replaced with scar tissue.
- Cardiomyopathy in Adults
- Pediatric Cardiomyopathies
- Dilated Cardiomyopathy
- Hypertrophic Cardiomyopathy
- Restrictive Cardiomyopathy
- Arrhythmogenic Right Ventricular Dysplasia
- Transthyretin Amyloid Cardiomyopathy
Cardiomyopathy is a disease of the heart muscle that makes it harder for the heart to pump blood to the rest of the body. The various types of the disease have many causes, signs and symptoms as well as treatments. In most cases, cardiomyopathy causes the heart muscle to become enlarged, thick or rigid. In rare instances, diseased heart muscle tissue is replaced with scar tissue.
As cardiomyopathy worsens, the heart becomes weaker. The heart becomes less able to pump blood throughout the body and incapable of maintaining a normal electrical rhythm. The result can be heart failure or irregular heartbeats called arrhythmias. A weakened heart also can cause other complications, such as heart valve problems.
Overview
The main types of cardiomyopathy are:
- Dilated cardiomyopathy
- Hypertrophic cardiomyopathy
- Restrictive cardiomyopathy
- Arrhythmogenic right ventricular dysplasia
- Transthyretin amyloid cardiomyopathy (ATTR-CM)
- Some other types of cardiomyopathy are called “unclassified cardiomyopathy.” Another type is “stress-induced cardiomyopathy,” also known as broken heart syndrome.
Cardiomyopathy can be “acquired” when it develops due to another disease, condition or factor. Or cardiomyopathy can be “inherited” when the gene for the disease is passed on from a parent.
In many cases, the cause of cardiomyopathy isn’t known. This is often the case when it occurs in children.
Cardiomyopathy affects all ages, although certain age groups are more likely to have certain types of cardiomyopathy.
Approaches to treatment
Some cases of cardiomyopathy have no signs or symptoms and need no treatment. In other cases, cardiomyopathy develops quickly with severe symptoms, and serious complications occur. Treatment is required in these instances.
Treatments include lifestyle changes, medications, surgery, implanted devices to correct arrhythmias and other nonsurgical procedures. These treatments can control symptoms, reduce complications and prevent the disease from worsening.
Cardiomyopathy is rare in children. For that reason alone, a diagnosis of cardiomyopathy can rattle parents, and possibly the child too. Fortunately, our understanding of how the heart works under normal and abnormal conditions is increasing each year.
As you familiarize yourself with pediatric cardiomyopathy, you’ll find yourself in a better position to evaluate your child’s treatment options. Knowledge is power. Learn all that you can as you work with your child’s doctor to identify the best course of action.
Cardiomyopathy and its incidence among children
Cardiomyopathy refers to a diseased state of the heart involving abnormalities of the muscle fibers, which contract with each heartbeat. It can be considered “primary” or “secondary”:
In primary cases, cardiomyopathy occurs because the muscle cells themselves are abnormal (usually due to a gene mutation).
Secondary cases of cardiomyopathy involve healthy heart muscle cells that are adversely affected by other conditions. Precipitating conditions include low blood flow to the heart, low blood oxygen, high blood pressure and certain infections.
According to the Pediatric Cardiomyopathy Registry, one in every 100,000 children in the U.S. under the age of 18 is diagnosed with cardiomyopathy. The majority of diagnosed children are under 12 months, followed by children 12 to 18 years old.
Types of cardiomyopathy
Cardiomyopathies can be grouped into four broad categories. The clinical features and treatment options differ for each.
- Dilated cardiomyopathy
- Hypertrophic cardiomyopathy
- Restrictive cardiomyopathy
- Miscellaneous (rare) cardiomyopathies
Dilated cardiomyopathy (DCM) is the most common type, occurring mostly in adults younger than 50. It affects the heart’s ventricles and atria, the lower and upper chambers of the heart.
Frequently, the disease starts in the left ventricle, the heart’s main pumping chamber. The heart muscle begins to dilate, stretching and becoming thinner. As a result, the inside of the chamber enlarges. The problem often spreads to the right ventricle and then to the atria.
As the heart chambers dilate, the heart muscle doesn’t contract normally and can’t pump blood very well. As the heart becomes weaker, heart failure can occur. Common symptoms of heart failure include shortness of breath, fatigue and swelling of the ankles, feet, legs, abdomen and veins in the neck.
Dilated cardiomyopathy also can lead to heart valve problems, arrhythmias (irregular heartbeats) and blood clots in the heart.
Other Names for Dilated Cardiomyopathy
- Alcoholic cardiomyopathy. (Overuse of alcohol causes the disease)
- Congestive cardiomyopathy
- Diabetic cardiomyopathy
- Familial dilated cardiomyopathy
- Idiopathic cardiomyopathy
- Ischemic cardiomyopathy (Coronary heart disease, also called coronary artery disease, or heart attack cause the disease. Not all forms of DCM are ischemic in origin.)
- Peripartum cardiomyopathy (When the disease develops in a woman shortly before or after she gives birth)
- Primary cardiomyopathy
What Causes Dilated Cardiomyopathy
The cause of dilated cardiomyopathy often isn’t known. Up to one-third of the people who have it inherit it from their parents.
Some diseases, conditions and substances also can cause the disease, such as:
- Coronary heart disease, heart attack, high blood pressure, diabetes, thyroid disease, viral hepatitis and HIV
- Infections, especially viral infections that inflame the heart muscle
- Alcohol, especially if you also have a poor diet
- Complications during the last month of pregnancy or within five months of birth
- Certain toxins such as cobalt
- Certain drugs (such as cocaine and methamphetamines) and two medicines that treat cancer (doxorubicin and daunorubicin)
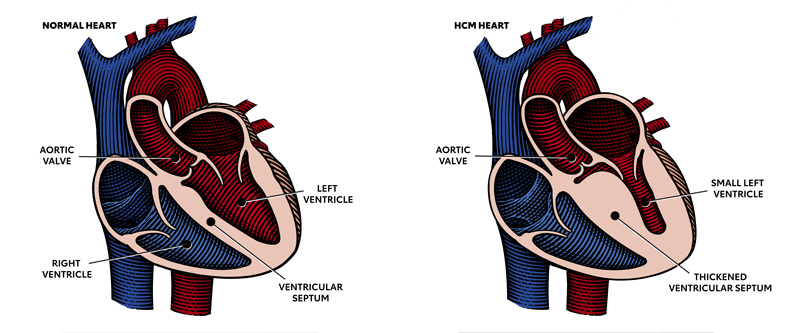
What is Hypertrophic Cardiomyopathy?
Hypertrophic cardiomyopathy is most often caused by abnormal genes in the heart muscle. These genes cause the walls of the heart chamber (left ventricle) to become thicker than normal.
The thickened walls may become stiff and this can reduce the amount of blood taken in and pumped out to the body with each heartbeat.
Restrictive cardiomyopathy tends to affect older adults. The heart’s ventricles become rigid because abnormal tissue, such as scar tissue, replaces the normal heart muscle. As a result, the ventricles can’t relax normally and fill with blood, and the atria become enlarged. Blood flow in the heart is reduced over time. This can lead to problems such as heart failure or arrhythmias.
Other Names for Restrictive Cardiomyopathy
- Idiopathic restrictive cardiomyopathy
- Infiltrative cardiomyopathy
What Causes Restrictive Cardiomyopathy
Certain diseases, conditions and factors can cause restrictive cardiomyopathy, including:
- Hemochromatosis: a disease in which too much iron builds up in your body. The extra iron is toxic to the body and can damage the organs, including the heart.
- Sarcoidosis: a disease that causes inflammation and can affect the body’s organs. Researchers believe that an abnormal immune response may cause sarcoidosis. The abnormal response causes tiny lumps of cells to form in the body’s organs, including the heart.
- Amyloidosis: a disease in which abnormal proteins build up in the body’s organs, including the heart.
- Connective tissue disorders
- Some cancer treatments, such as radiation and chemotherapy
Arrhythmogenic right ventricular dysplasia (ARVD) is a rare type of cardiomyopathy. It occurs when the muscle tissue in the right ventricle dies and is replaced with scar tissue. This disrupts the heart’s electrical signals and causes arrhythmias. Symptoms include palpitations and fainting after physical activity. Palpitations are feelings that your heart is skipping a beat, fluttering or beating too hard or too fast.
ARVD usually affects teens or young adults. It can cause sudden cardiac arrest (SCA) in young athletes.
Other Names for ARVD
- Arrhythmogenic right ventricular cardiomyopathy
- Right ventricular cardiomyopathy
- Right ventricular dysplasia
What Causes ARVD
- Researchers believe that ARVD is an inherited disease.
What is transthyretin amyloid cardiomyopathy?
Transthyretin (trans-thy-re-tin) amyloid cardiomyopathy (ATTR-CM) is an underdiagnosed and potentially fatal disease of the heart muscle. In ATTR-CM, a protein called transthyretin that normally circulates in the bloodstream becomes misshapen and builds up in the heart, nerves and other organs.
When these amyloid deposits build up in the heart, the walls can become stiff, making the left ventricle unable to properly relax and fill with blood – called cardiomyopathy. As the condition progresses, the heart can become unable to adequately squeeze to pump blood out of the heart, ultimately leading to heart failure.
Hereditary ATTR-CM
There are two types of ATTR-CM. In hereditary ATTR-CM (hATTR-CM), which can run in families, there’s a variant in the transthyretin gene, which results in amyloid deposits in the heart, nerves and sometimes the kidneys and other organs. Symptoms may start as early as age 20 and as late as 80.
Hereditary ATTR-CM is more common in localized parts of Portugal, Sweden and Japan; however, there are a number of variants in different parts of the world. Some variants are more common in people of Irish ancestry while others are common among people of African descent.
Different variants may progress in a different way and involve different organs. The most common variant in the United States occurs in 1 in 25 of all African Americans and in older patients who may be misdiagnosed with high blood pressure-related heart disease.
Genetic testing may provide important information to developing a treatment plan.
Wild-type ATTR-CM
The second type is wild-type ATTR-CM (wATTR-CM), in which there is no variant in the transthyretin gene. Wild-type ATTR-CM doesn’t run in families. It most commonly affects the heart and can also cause carpal tunnel syndrome and pain and numbness in the hands and feet, called peripheral neuropathy. Symptoms usually start after age 65.
What are the risk factors?
Risk factors for hereditary ATTR-CM include:
- A family member with ATTR-CM or heart failure
- Age 50 and older (although symptoms can begin anywhere from age 20 to 80)
- Gender (patients are primarily male)
- Race – African American
Risk factors for wild-type ATTR-CM include:
- Age 65 and older
- Gender (patients are primarily male)
What are the symptoms?
Symptoms of ATTR-CM can vary or be subtle, and the condition is often misdiagnosed. In its early stages, it may mimic the symptoms of other conditions, such as heart failure related to high blood pressure, or hypertension, and enlargement and thickening of the heart, or hypertrophic cardiomyopathy. Some patients may have no symptoms, while others may progress to end-stage heart failure. The symptoms of wild-type ATTR-CM may be mild and remain undiagnosed.
ATTR-CM symptoms are like those associated with heart failure.
Shortness of breath is the most common, especially with minimal exertion and when lying down.
Other symptoms usually occur after the shortness of breath is already there, including:
- Coughing or wheezing, especially when lying down.
- Swelling in the feet, ankles and legs
- Bloating in the abdomen
- Confusion or trouble thinking
- Increased heart rate
- Palpitations or abnormal heart rhythms
Additional symptoms for ATTR-CM may include:
- Numbness or tingling in the hands and feet (hATTR-CM)
- Carpal tunnel syndrome (wATTR-CM)
How is it diagnosed?
ATTR-CM may be suspected because of typical symptoms and the results of a routine cardiac test — an electrocardiogram or echocardiogram. Once suspected, more specialized tests are needed to confirm the diagnosis. These could include:
- Imaging studies of the heart, most commonly a cardiac MRI and/or a nuclear medicine scan of the heart
- A tissue biopsy of an affected organ
- Genetic testing
How can ATTR-CM be treated?
There are several promising new therapies for ATTR-CM on the horizon or available, so it is important to talk to your health care professionals about treatment options.
With ATTR-CM, health care professionals focus on easing the heart failure symptoms and slowing or stopping the amyloid deposits. Medications are approved for hereditary transthyretin amyloidosis affecting the nerves, causing a condition called neuropathy.
In cases of advanced heart failure, heart transplantation may be an option. Because the abnormal transthyretin protein is produced by the liver, some patients may require both heart and liver transplantation.
Talk with your health care professionals
Awareness of ATTR-CM among health care teams is low. In fact, it’s often misdiagnosed as hypertensive heart failure or hypertrophic cardiomyopathy and may already be advanced by the time the patient receives a diagnosis.
If you are living with heart failure and have additional unresolved or seemingly unrelated symptoms, speak to your health care professional about your symptoms and treatment options. Starting that conversation could be a lifesaver.
Tell your doctor about any shortness of breath and ask for a diagnostic testing and treatment plan. If the diagnostic tests don’t provide an answer, or if the treatment fails to improve your symptoms, talk with your health care professional about next steps. It’s important not to give up until the tests reveal a diagnosis and the treatment relieves your symptoms.
If you do have ATTR-CM, genetic testing may offer important information to your health care professional to develop a treatment plan.
Get support
Getting support for you and your caregivers is important to your health. Research shows that going it alone and not seeking support during a heart diagnosis can reduce your ability to make the lasting changes you need to live a longer, healthier life.
Learn more about ATTR-CM and connect with others impacted by this form of cardiomyopathy on the American Heart Association’s Support Network. There, you will find a community triumphing over their health obstacles each day.
You can share your experience to help others and get support from others who have been there. Connect for free today at heart.org/SupportNetwork.